 A “fast-track” medical device approval program established by the Food and Drug Administration (FDA) in the 1980s is the subject of updated guidelines announced on July 28, 2014. The 510(k) program has been criticized in the past for its alleged laxity. The program was originally instituted to speed the approval of basic medical items like Band-Aids and tongue suppressors. A manufacturer could be exempted from clinical trials and other costly tests by claiming that a newer device was “substantially equivalent” to the predicate device. This expedited the device’s arrival in the marketplace in Mississippi and elsewhere while simultaneously saving both the regulators and the manufacturers time and money.
A “fast-track” medical device approval program established by the Food and Drug Administration (FDA) in the 1980s is the subject of updated guidelines announced on July 28, 2014. The 510(k) program has been criticized in the past for its alleged laxity. The program was originally instituted to speed the approval of basic medical items like Band-Aids and tongue suppressors. A manufacturer could be exempted from clinical trials and other costly tests by claiming that a newer device was “substantially equivalent” to the predicate device. This expedited the device’s arrival in the marketplace in Mississippi and elsewhere while simultaneously saving both the regulators and the manufacturers time and money.
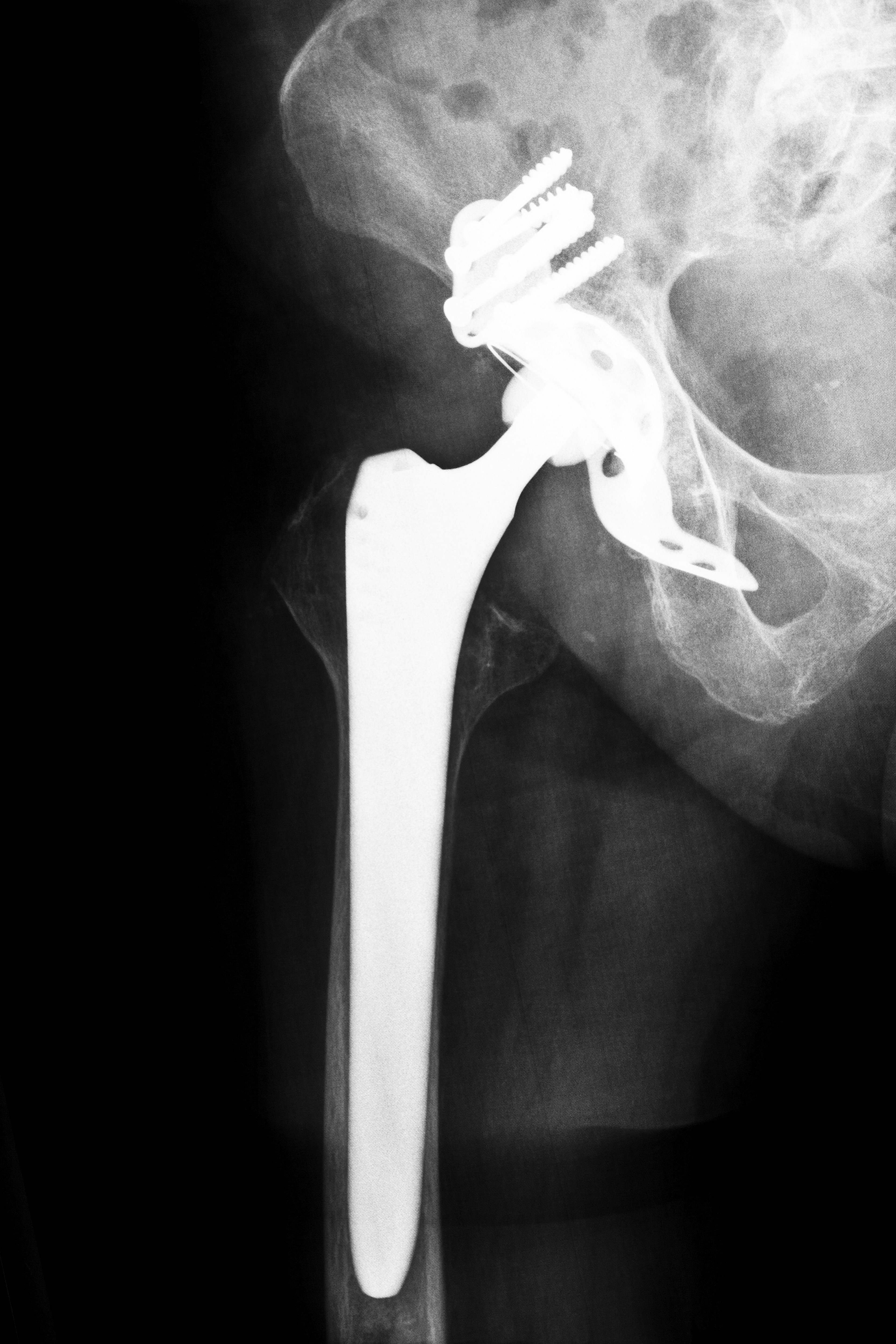
Later, the use of the program expanded into the realm of more complicated medical devices. Eventually, manufacturers were claiming that their artificial hip joints, defibrillators and other complex devices were “substantially equivalent” to earlier versions. Testing requirements were waived, and in some cases, devices were used on Mississippi patients and others across the country who are now involved in defective medical device lawsuits.
This alleged abuse of the 510(k) program has caused some, like Institute of Medicine Chairman Dr. David Challoner, to call for an end to the program. In 2011, Dr. Challoner asserted the view that the FDA had been too often persuaded by the medical device industry to promote expediency rather than public safety in the discharge of its duties. He alleged that the shortcomings of certain vital devices had been discovered only after they had been used in numerous patients. These allegedly defective medical devices have generated many lawsuits filed on behalf of patients in Mississippi and across the country who suffered as a result.
The new guidelines published by the FDA on July 28, 2014, seek to more clearly specify how and when substantial equivalency is to be asserted. The FDA now requires that a new medical device be used in the very same way that the predicate device was used, and the agency requires that it have the very same technological features that the predicate device possessed. If there are technological updates or alterations, the manufacturer must openly address any safety concerns that these changes may engender. Scientific data or the results of clinical trails need to be submitted to support the manufacturers’ contentions.
Injured by a Defective Medical Device?
Regardless of the potential success of these new FDA guidelines, some defective medical devices have already entered the marketplace for many years under the 510(k) program. Some of these devices, such as certain types of transvaginal surgical mesh and certain artificial joints, have been alleged to be defective. If you or a loved one has been adversely affected by a defective medical device, Attorney Group for Mississippi is available to discuss your case. Please contact us today so that we can help answer your questions and connect you with an affiliated attorney who can assist with your case. Contact us for a free consultation to learn more.

{kind=link}